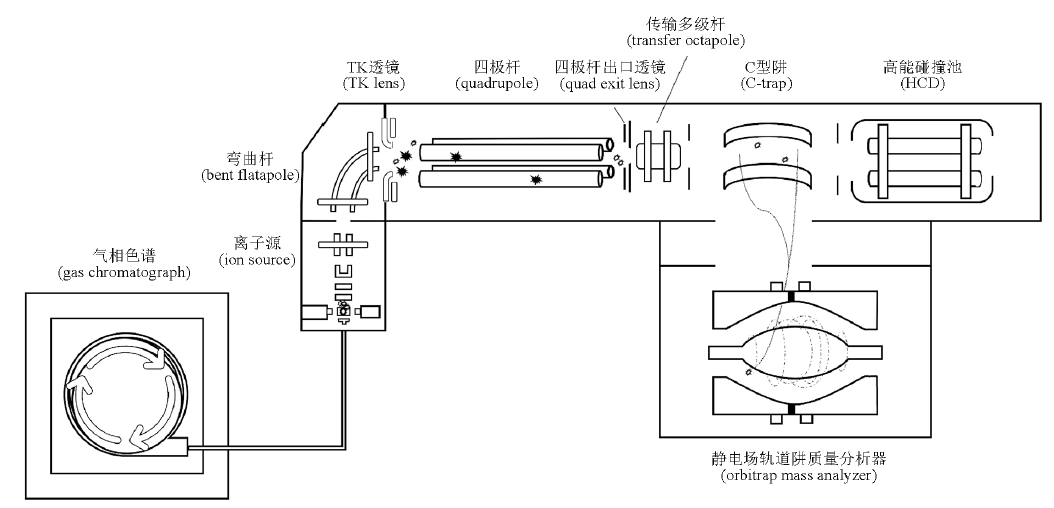
李雅静, 张振凌, 王胜超, 朱建光, 王瑞生, 李宝卿, 赵永琪, 孙梦梅
目的:基于指纹图谱和网络药理学方法,对曼地亚红豆杉质量标志物(Q-Marker)进行预测,建立基于Q-Marker的曼地亚红豆杉药材评价方法。方法:采用Waters SymmmetryShieldTM RP 18(250 mm×4.6 mm,5 μm)色谱柱,以乙腈-0.1%三氟乙酸水为流动相,梯度洗脱,流速1 mL·min-1,柱温30 ℃,检测波长254 nm,进样体积10 μL,建立8个年限24批曼地亚红豆杉药材指纹图谱并标定共有峰,采用层次聚类分析(hierarchical clustering analysis, HCA)进行分类评价,借助正交偏最小二乘法-判别分析(orthogonal partial least square-discriminant analysis, OPLS-DA)筛选不同年限曼地亚红豆杉的主要差异性标志物;结合网络药理学,通过相应数据库筛选差异性标志物的核心靶点和关键通路,构建“成分-靶点-通路”网络图,以此预测曼地亚红豆杉的Q-Marker;采用HCA及主成分分析(principal component analysis, PCA)进一步验证曼地亚红豆杉的Q-Marker,综合评价其质量。结果:建立了24批曼地亚红豆杉药材指纹图谱,标定了25个共有峰,指认出18个化合物,包括紫杉烷类、黄酮类、生物碱类、甾体类、酚类,相似度均>0.900;HCA的结果显示不同年限曼地亚红豆杉存在一定的差异性;经OPLS-DA筛选出11个差异性标志物,分别为紫杉醇、10-脱乙酰基巴卡亭Ⅲ(10-DAB)、巴卡亭Ⅲ、三尖杉宁碱、10-去乙酰紫杉醇(10-DAT)、阿魏酸、山柰素、芦丁、金松双黄酮、穗花杉双黄酮、对羟基苯甲醛;基于差异性标志物,运用网络药理学从有效性角度对其进行分析,初步预测紫杉醇、10-DAB、巴卡亭Ⅲ、三尖杉宁碱、10-DAT为曼地亚红豆杉的Q-Marker,主要通过作用于PIK3R1、AKT1、EGFR、HRAS、MAPK1等14个核心靶点,调控癌症通路、AGE-RAGE信号在糖尿病并发症中的作用等信号通路,发挥消肿散结、通经利尿的功效;HCA结果验证了预测的Q-Marker的合理性,PCA综合评价结果表明十年生春季采收的曼地亚红豆杉质量较佳。结论:基于指纹图谱和网络药理学方法筛选出紫杉醇、10-DAB、巴卡亭Ⅲ、三尖杉宁碱、10-DAT为曼地亚红豆杉的Q-Marker,结合化学识别模式方法对不同年限曼地亚红豆杉的质量进行综合排序,其中十年生春季采收的得分最高,质量较好,为曼地亚红豆杉的质量标准的研究及资源评价提供了新思路。