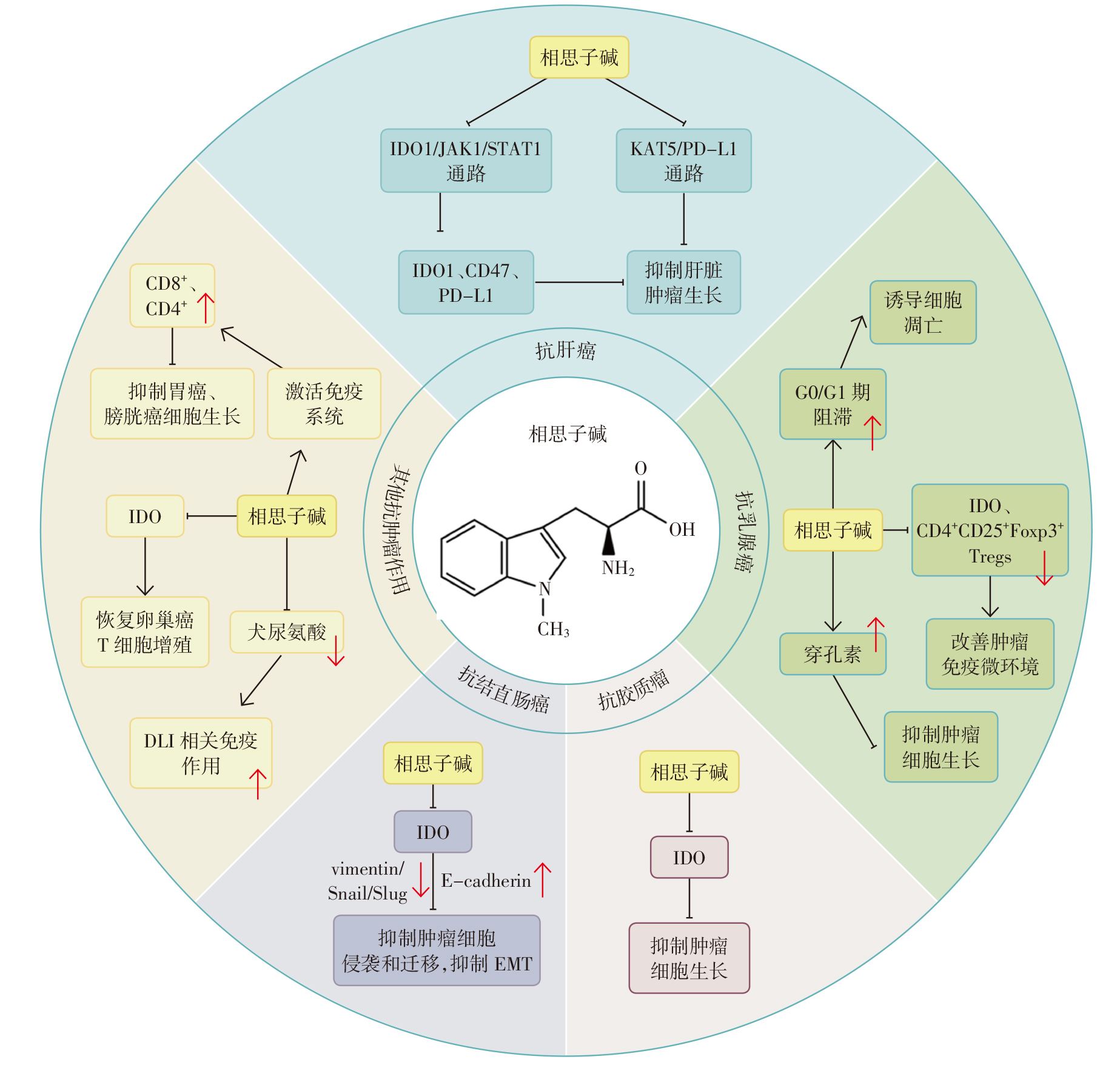
周国梁, 俞浩, 陈浩, 王钰, 尚尔鑫, 宿树兰, 段金廒
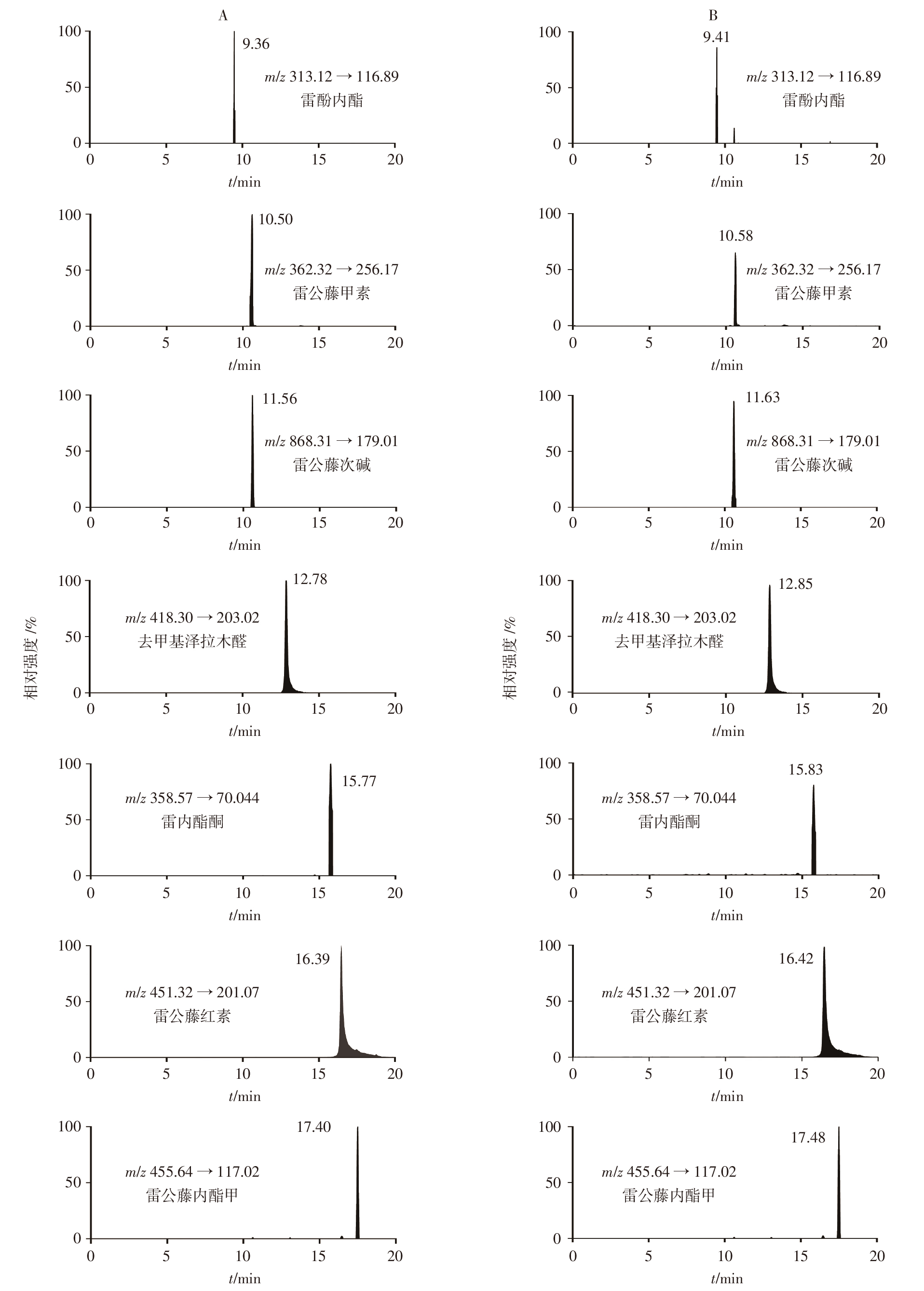
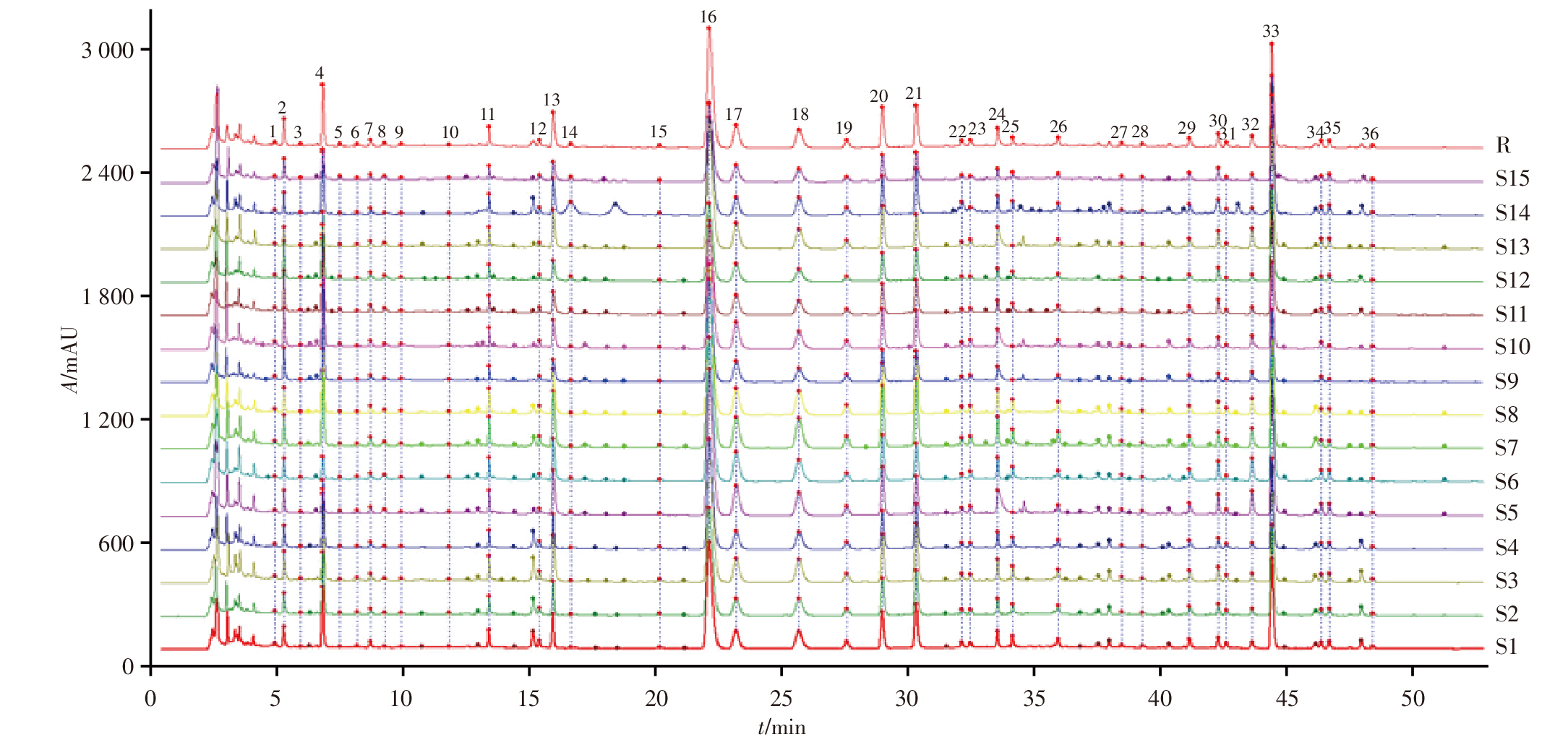
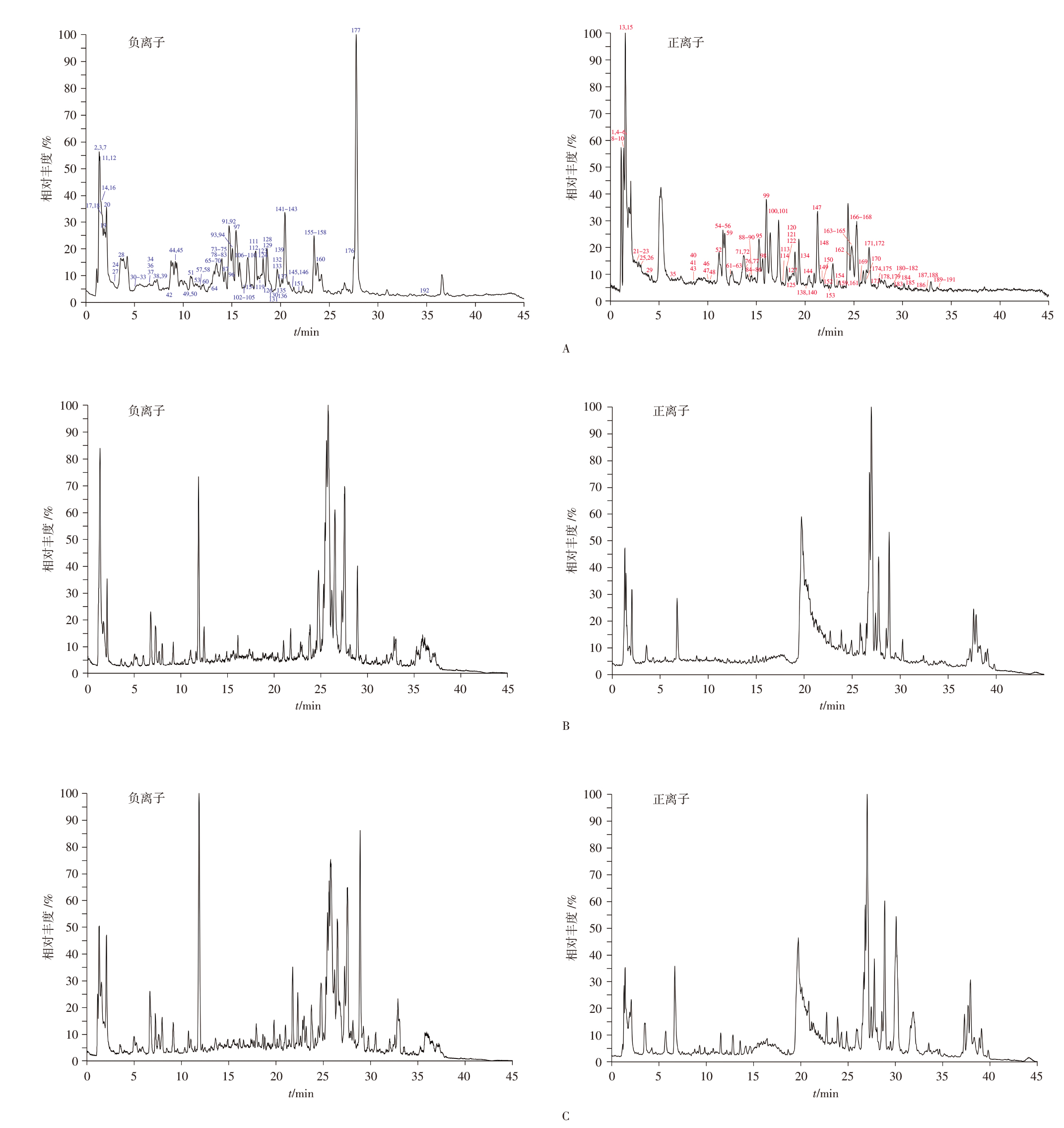
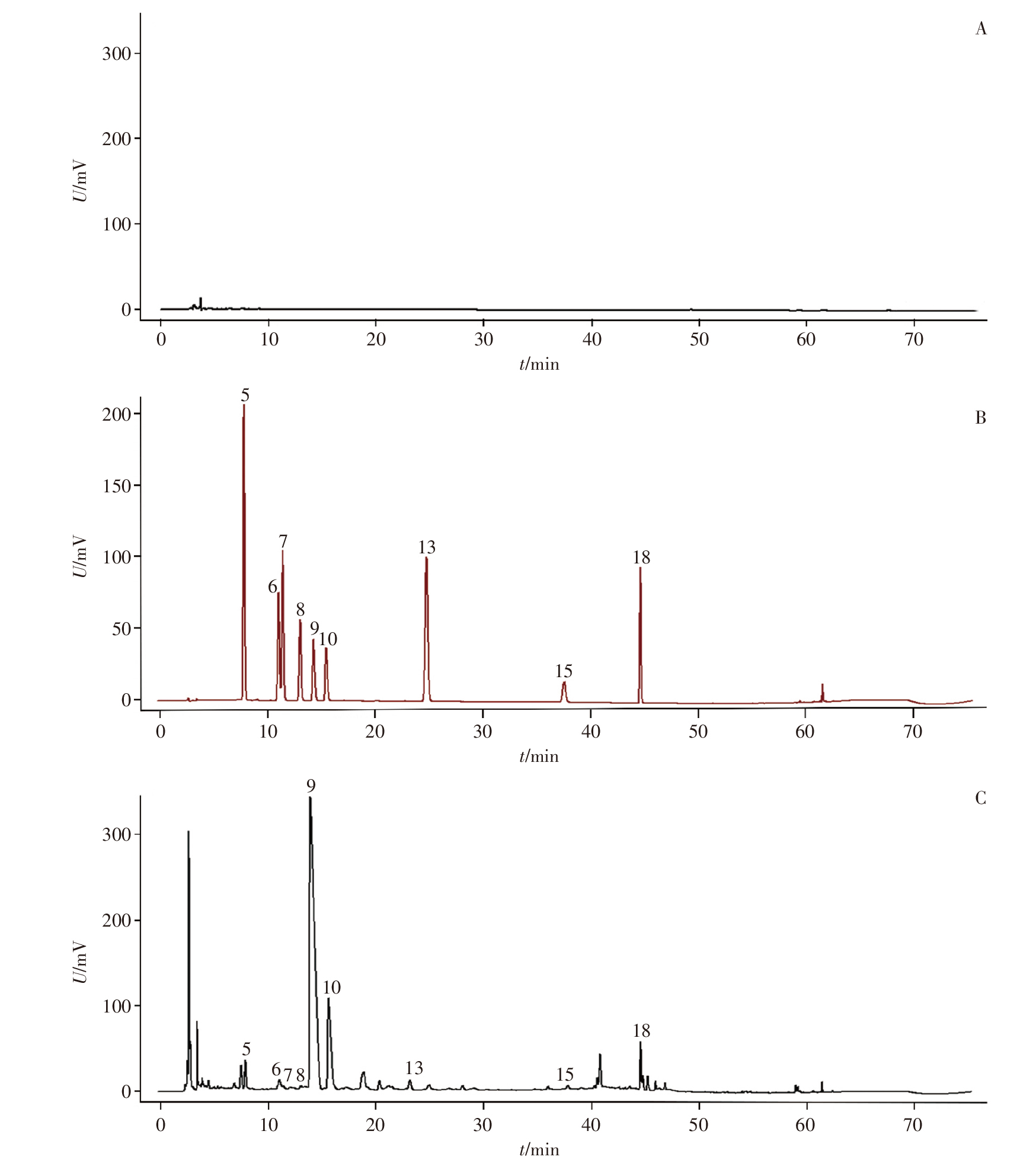
目的: 建立超高效液相色谱-三重四极杆质谱(UPLC-TQ/MS)联用法同时测定雷公藤与凤尾草中13个化学成分的含量,对雷公藤-凤尾草不同配伍比例下化学成分的溶出变化进行定量分析;从化学成分角度分析成分的溶出变化,确定最优配伍比例,为进一步阐明量效关系和指导临床用药提供依据。方法: 以水和70%乙醇提取不同配伍比例(1 ∶ 4、1 ∶ 2、1 ∶ 1、2 ∶ 1、4 ∶ 1)雷公藤-凤尾草中的成分,应用UPLC-TQ/MS联用技术,对不同配伍比例组中13个化学成分的溶出度进行分析。采用AcquityTM UPLC BEH C18色谱柱(50 mm×2.1 mm,1.7 μm),以乙腈-0.1%甲酸水溶液为流动相进行梯度洗脱,流速为0.3 mL · min-1,采用UPLC-TQ/MS电喷雾正、负离子源(ESI+/ESI-),在多反应监测(MRM)模式下进行检测。结果: 雷公藤与凤尾草以不同比例配伍后,经70%乙醇提取测定发现,不同配伍比例组中6个雷公藤萜类成分的总溶出度均低于70%乙醇雷公藤单煎组(C0组),且有显著性差异(P<0.05);不同比例配伍的水提取物组中,6个雷公藤萜类成分的总溶出度亦低于70%乙醇提取物组,尤其是雷公藤与凤尾草1 ∶ 2配伍的水提组(S2组),溶出度最低。在雷公藤次碱的提取分析中,雷公藤与凤尾草不同比例配伍后,无论是水提取还是70%乙醇提取,各配伍比例组的溶出度均显著高于水提取雷公藤单煎组(S0组)和C0组,且有极显著差异(P<0.01),表明雷公藤与凤尾草配伍可促进雷公藤次碱的溶出,其中以S2组溶出度最高。在对凤尾草中黄酮类成分的分析中,不同比例配伍后经70%乙醇提取,凤尾草中6个黄酮类成分的总溶出度低于70%乙醇凤尾草单煎组(FC0组),且存在显著性差异(P<0.05);而不同配伍组经水提取后,凤尾草中6个黄酮类成分的总溶出度高于70%乙醇提取组,且与水提凤尾草单煎组(FS0)相比有显著性差异(P<0.01),其中S2组中6个凤尾草黄酮类成分的总溶出度最高。结论: 雷公藤与凤尾草在不同配伍比例下,水提取和70%乙醇提取对6个雷公藤萜类成分、雷公藤次碱及6个凤尾草黄酮类成分的溶出度均有显著影响,其中以S2组效果最为显著,为临床安全用药提供了科学依据。