陈娟, 焦阳, 汪冰, 王琳, 薛菲, 解盈盈, 闫庆康, 李向, 林永强
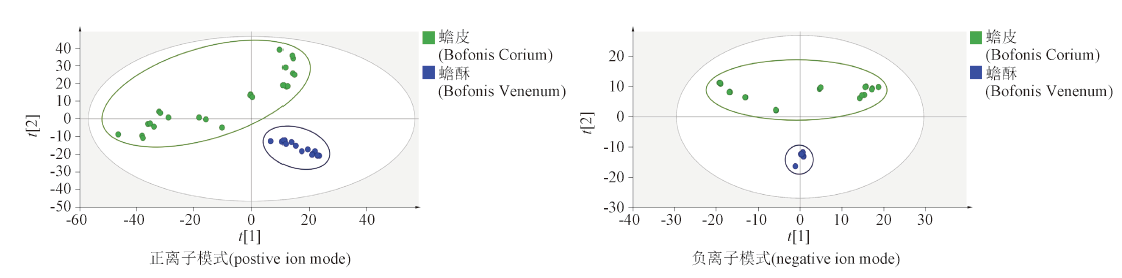
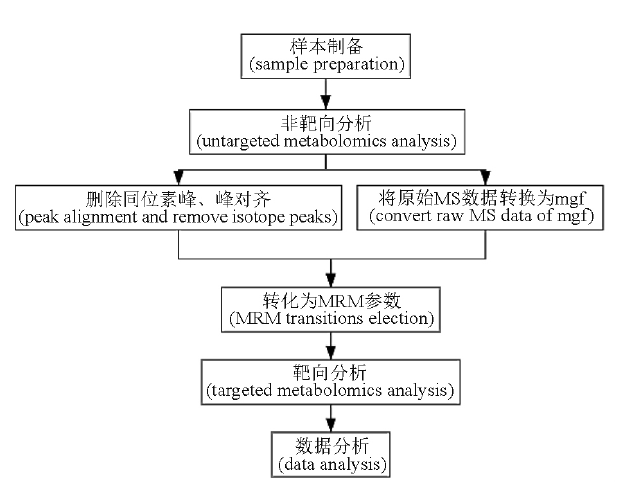
目的: 应用超高效液相色谱-四极杆-静电场轨道阱高分辨质谱(UPLC-Q-Exactive Orbitrap MS)结合化学计量学对蟾皮区别于蟾酥的差异成分进行研究和应用。方法: 采用Waters Atlantis T3(150 mm×2.1 mm,3 μm)色谱柱,以乙腈-0.1%甲酸溶液为流动相,进行梯度洗脱,流速0.3 mL·min-1,柱温30 ℃,选用UPLC-Q-Exactive Orbitrap MS分别在正、负离子模式下采集数据,通过主成分分析(PCA)和正交偏最小二乘-判别分析(OPLS-DA)得到候选差异离子,通过Xcalibur 3.0数据处理系统提取差异离子的二级碎片信息,再使用AB SCIEX 6500+三重四极杆质谱的多反应监测模式(MRM)对候选差异离子进行专属性验证,应用专属性良好的离子对建立蟾酥中掺伪蟾皮的鉴别方法,并进行方法学考察。结果: 经查找验证,共找到5个可用于识别蟾皮的差异离子对(m/z: 377.5→243.2,172.1;330.4→170.9,127.0;313.4→201.2,171.0;452.4→280.2,298.2;614.7→332.4,281.3),应用差异离子信息建立了蟾酥中蟾皮的检查方法并进行了方法学考察。考察结果表明,所建方法具有较好的专属性、耐用性,各离子对在一定范围内均呈现良好的线性关系,相关系数均大于0.996 7,精密度试验RSD为1.3%~4.1%,重复性试验RSD为1.3%~3.2%,依据上述方法对5批蟾酥市场样品进行测定,结果有2批样品出现蟾皮成分但未超出拟定的5%掺伪上限。结论: 本研究报道了一种将UPLC-Q-Exactive Orbitrap MS和化学计量学相结合的快速寻找差异信息的方法,并应用差异信息建立了蟾酥中掺伪蟾皮的检查方法。