姜丹凤, 黄京秋, 吴奇珍, 王漪璇, 方雅玲, 吴维怡, 吴文英, 孙艳
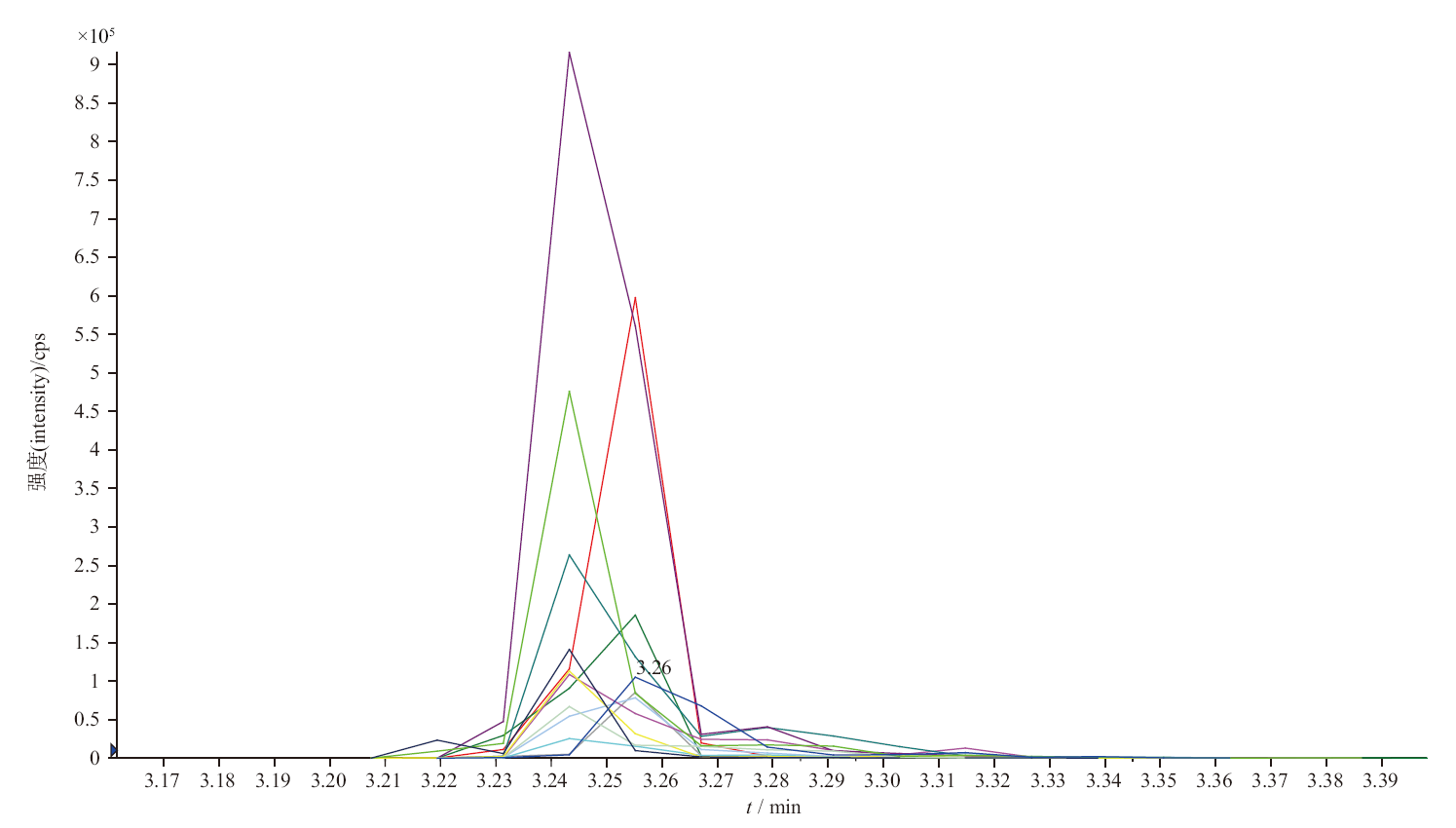
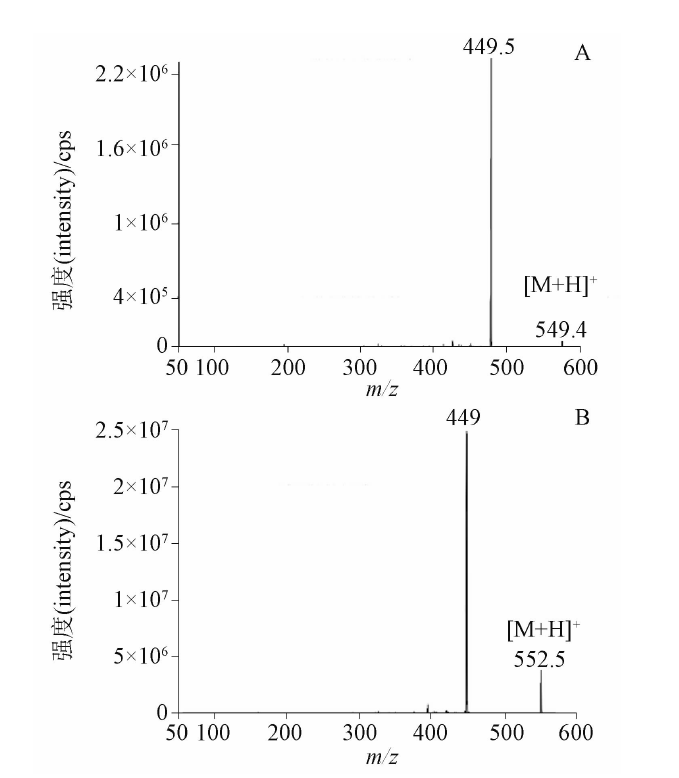
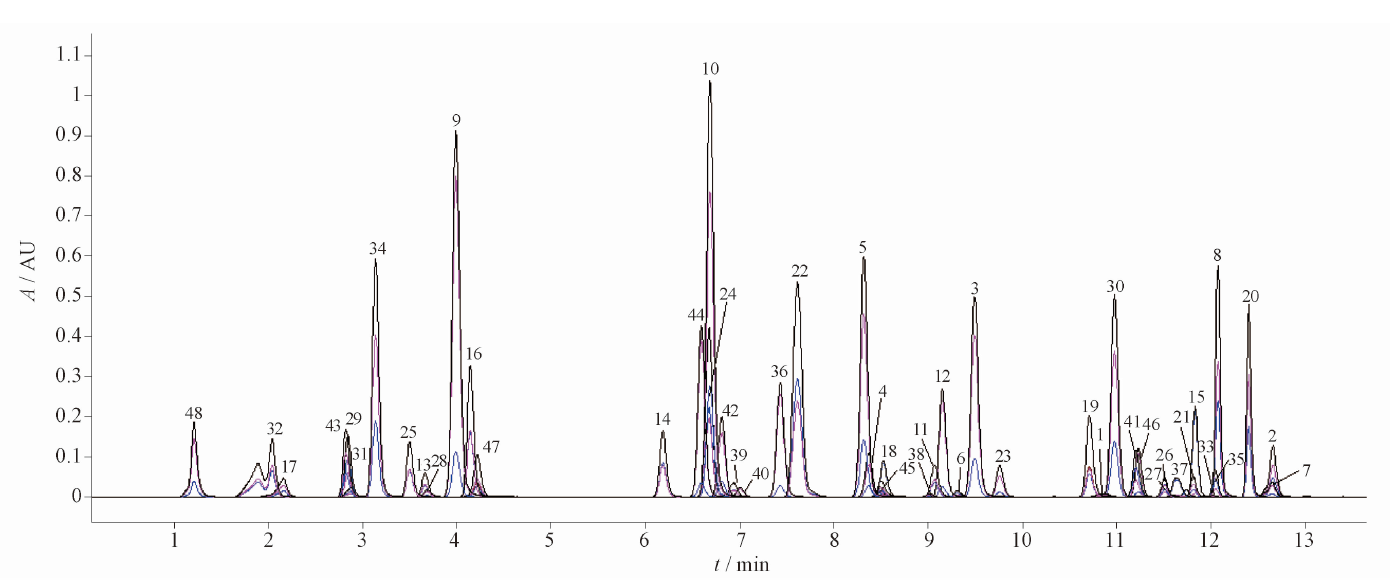
目的: 建立测定人血浆中FCN-437c药物浓度的HPLC-MS/MS方法,并用于FCN-437c的 Ⅰ 期临床研究。方法: 血浆经蛋白沉淀处理后,采用HPLC-MS/MS法进样测定。色谱柱为YMC Triart PFP柱(50 mm×2.1 mm, 5 μm),以0.5%甲酸水溶液(含5 mmol·L-1乙酸铵,A)-乙腈(B)为流动相,梯度洗脱,流速为0.5 mL·min-1,柱温为35 ℃,进样量为2 μL,进样器温度为10 ℃。质谱采用ESI+,MRM模式。检测离子反应对为m/z 549.4→449.5(FCN-437c)和m/z 552.5→449.0(FCN-437-D3,氘代内标),雾化气压力276 kPa,辅助气压力207 kPa,去簇电压100 V,碰撞室出口电压25 V。结果: 人血浆中FCN-437c的线性范围为5~1 000 ng·mL-1(r=0.999 0),定量限为5 ng·mL-1,批内、批间精密度分别小于2.0%和4.1%,平均回收率为104.0%(FCN-437c)、78.6%(FCN-437-D3),内标归一化基质因子为100%~102%。FCN-437c储备液4 ℃放置202 d,FCN-437c及FCN-437-D3工作液室温放置24 h,血浆样品室温放置20 h、冻融四循环、-20 ℃放置134 d、-80 ℃放置662 d,样品处理后自动进样器放置24 h,全血样品室温放置4 h均稳定。应用此方法检测了受试者口服FCN-437c后血浆药物浓度,ISR样品测试结果为97.0%与初测值的偏差在±20%以内。血浆样本稀释10倍后准确度为102.0%~108.0%。FCN-437c连续给药与单次给药相比,RAUC0-24和RCmax的累积比为1.33倍与1.59倍。结论: 本方法简便、准确、耐用、专属性强,可满足人血浆中FCN-437c的定量分析的要求。