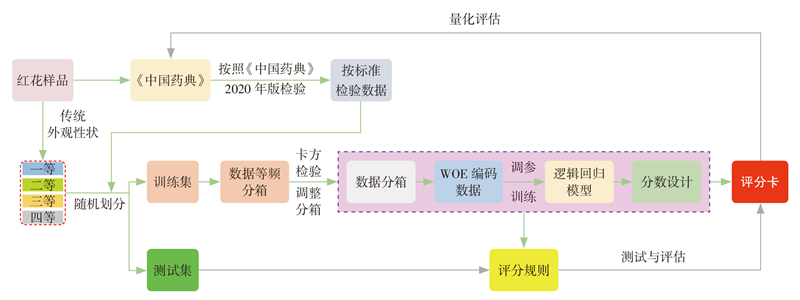
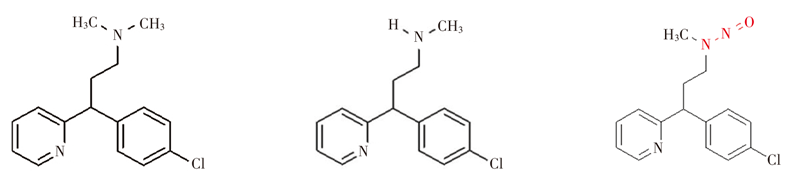
郑含笑, 李梦雨, 蒲婧哲, 胡冲, 陈灵丽, 张亚中
目的: 探讨不同生长年限暗紫贝母中糖类成分动态变化规律,为暗紫贝母的规范栽培、采收及质量控制提供科学依据。方法: 采用硫酸-蒽酮法测定暗紫贝母多糖含量,检测波长625 nm;采用高效液相色谱-紫外检测器法(HPLC-UV法)对暗紫贝母多糖的单糖组成及含量进行分析,多糖经酸水解后采用PMP进行柱前衍生化处理,使用Waters Symmetry C18色谱柱(250 mm×4.6 mm,5 μm),柱温35 ℃,以乙腈-0.02 mol · L-1乙酸铵(17 ∶ 83)为流动相,流速1 mL · min-1,检测波长245 nm,进样量5 μL;采用高效液相色谱-蒸发光散射检测器法(HPLC-ELSD法)对暗紫贝母中蔗糖和淀粉含量进行分析,使用Alltech chrom Prevail Carbohyrate ES色谱柱(250 mm×4.6 mm,5 μm),柱温35 ℃,以乙腈-水(75 ∶ 25)为流动相,流速1 mL · min-1,蒸发光散射检测器漂移管温度110 ℃,气体流量2.5 L · min-1,进样量5 μL。结果: 不同生长年限暗紫贝母中糖类成分含量存在显著差异。多糖含量均值呈“先升-后降-再上升”的动态变化趋势,2年生的多糖含量为5.46 mg · g-1,3年生的升至6.04 mg · g-1,4年生的略降至5.14 mg · g-1,5年生的显著上升至8.10 mg · g-1。在多糖的水解产物中,葡萄糖为主要单糖,其占比随生长年限增加而显著升高,范围为58.84%~65.77%;鼠李糖、半乳糖与阿拉伯糖含量相对较低,分别波动于0.74%~0.94%、1.26%~1.50%与1.33%~1.5%,未呈显著趋势。蔗糖含量均值亦表现出动态积累特征,2年生的蔗糖含量均值为54.54 mg · g-1,3年生的升至64.28 mg · g-1,4年生的略降至58.59 mg · g-1,5年生的则迅速上升至97.60 mg · g-1,呈现“前期波动、后期迅速积累”的趋势。淀粉含量均值在2、3、4年生阶段逐年升高,分别为178.42、197.61、233.51 mg · g-1,于4年生的淀粉含量均值达峰值,5年生的则略降至216.55 mg · g-1,表现为“逐年增加后略有回落”的趋势。结论: 不同生长年限的暗紫贝母中糖类成分含量存在显著差异,且随年限延长呈规律性变化。5年生的鳞茎中多糖和蔗糖含量最高,4年生的为淀粉积累高峰,葡萄糖为主要单糖且其含量随年限增加而上升。建议将蔗糖、淀粉及多糖作为暗紫贝母质量评价的重要指标,为其采收年限的优化与质量控制体系的建立提供科学依据。