邓丽华, 许克宁, 袁晓梅, 丁兵, 王丽苹, 徐丽, 杨本官, 刘源慧, 范建伟
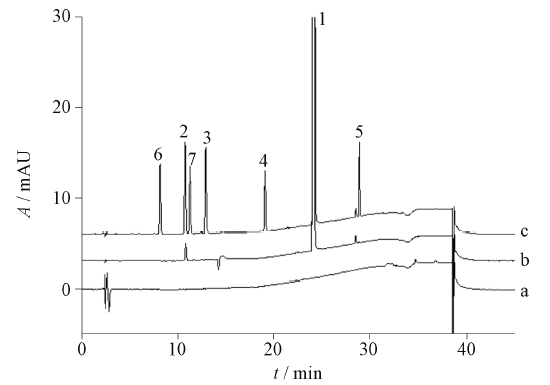
目的: 建立同时测定金藤清痹颗粒中青藤碱、新绿原酸、木兰花碱、隐绿原酸、绿原酸、异绿原酸B、异绿原酸A、异绿原酸C、哈巴俄苷、藁本内酯和甘草酸铵11个成分含量的UHPLC法,并结合主成分分析对制剂进行质量评价。方法: 采用UHPLC波长切换法,色谱柱为Waters XSelect® CSH C18柱(150 mm×4.6 mm,2.5 μm),以甲醇-乙腈(1∶1)为流动相A,0.2%磷酸水溶液为流动相B,梯度洗脱,流速1.0 mL·min-1,柱温25 ℃,检测波长218 nm(0~17 min时,检测青藤碱)、326 nm (17~25 min和34~98 min时,检测新绿原酸、隐绿原酸、绿原酸和异绿原酸A、异绿原酸B、异绿原酸C)、263 nm(25~34 min和98~125 min时,检测木兰花碱、哈巴俄苷、藁本内酯、甘草酸铵)。利用SPSS27.0软件对20批金藤清痹颗粒中成分含量进行多元统计分析。结果: 青藤碱、新绿原酸、木兰花碱、隐绿原酸、绿原酸、异绿原酸B、异绿原酸A、异绿原酸C、哈巴俄苷、藁本内酯和甘草酸铵11个成分的质量浓度分别在14.36~143.61 μg·mL-1(r=0.999 9)、7.71~77.15 μg·mL-1 (r=0.999 8)、9.18~91.83 μg·mL-1(r=0.999 7)、10.71~107.07 μg·mL-1(r=0.999 8)、12.88~128.80 μg·mL-1(r=0.999 8)、5.20~51.95 μg·mL-1(r=0.999 7)、5.18~51.84 μg·mL-1(r=0.999 8)、5.40~53.95 μg·mL-1(r=0.999 8)、2.62~26.16 μg·mL-1(r=0.999 9)、6.31~63.06 μg·mL-1(r=0.999 9)和11.13~111.26 μg·mL-1(r=0.997 6)范围内与峰面积的线性关系良好;平均加样回收率(n=6)分别为98.3%、98.3%、98.5%、98.9%、99.2%、101.0%、98.1%、97.1%、96.8%、98.0%和98.7%,RSD均小于3.0%。20批金藤清痹颗粒样品中,上述青藤碱等11个成分的含量测定结果(n=6)依次为2.206~2.704、1.071~1.403、2.096~2.487、1.321~1.724、2.241~2.612、0.605~0.749、0.363~0.412、0.835~1.020、0.151~0.191、0.791~1.188和1.008~1.363 mg·g-1。主成分分析结果显示,连续生产的金藤清痹颗粒批次间质量差异较小,且以样品S1、S5和S7的综合质量相对更好。结论: 建立的UHPLC多指标成分含量测定方法简便、准确、稳定,结合主成分分析,可全面地评价金藤清痹颗粒的产品质量。