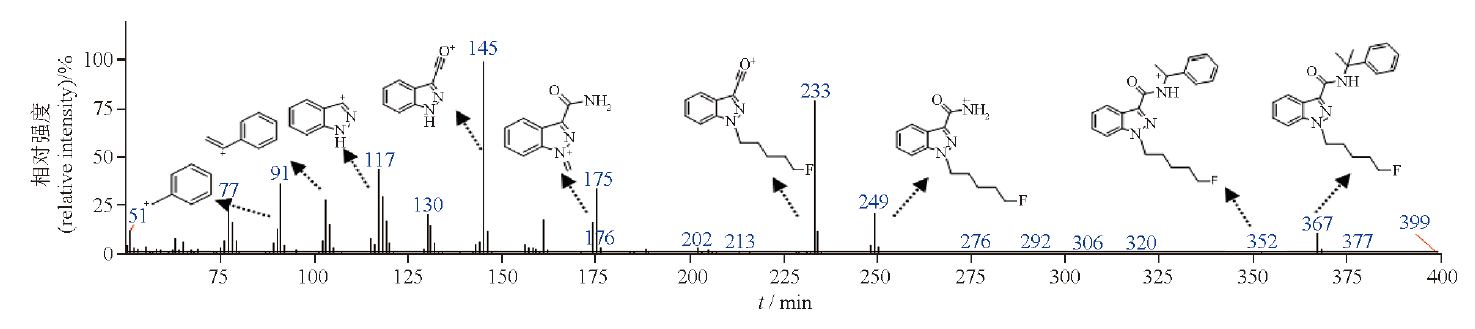
李高岩, 杨一荻, 袁鹤飞, 曲范娜, 李慧勇, 孙磊, 马双成, 笔雪艳
目的: 建立同时检测板蓝根中6个成分(尿苷、腺嘌呤、鸟苷、(R,S)-告依春、腺苷、直铁线莲宁B)的HPLC方法,考察双标线性校正(LCTRS)法对于板蓝根多成分定性分析的可行性。方法: 采用HPLC法,以甲醇为流动相A,水为流动相B,梯度洗脱(0~3 min,3%A; 3~18 min, 3%A→14%A; 18~25 min, 14%A→26%A; 25~34 min, 26%A; 34~40 min, 26%A→46%A; 40~60 min, 46%A→90%A),流速0.8 mL·min-1,柱温30 ℃,检测波长为0~32 min,254 nm;32~60 min,230 nm,进样量10 μL;测定板蓝根中6个成分在16根不同品牌和型号的C18色谱柱的实际保留时间,以鸟苷和直铁线莲宁B 2个成分作为双标化合物,采用LCTRS法定位各成分色谱峰,并用3根未知色谱柱进行方法验证。同时以鸟苷为参照物,采用RRT法预测其他5个成分的保留时间。比较这2种方法的预测准确性和色谱柱符合率。结果: LCTRS法能够较好地对6个指标性成分进行保留时间的预测和定性分析。与RRT法相比较,LCTRS法所预测结果准确性更高,色谱柱普适性更优。结论: LCTRS法用于同时测定板蓝根中多成分可行且准确,操作简便,耐用性好,具有推广价值。